The jiggling and wiggling of atoms in energy materials
Energy materials are the key ingredients to convert the sun’s energy into electrical energy and store it in batteries. Yet predicting the properties of these materials under technologically relevant conditions has been a major challenge for researchers. The team led by David Egger develops new theoretical methods in order to discover energy materials and improve devices made from them.

Focus Group: Theory of Functional Energy Materials
Prof. David Egger (TUM), Alumnus Rudolf Mößbauer Tenure Track Assistant Professor | Dr. Sebastián Caicedo-Dávila (TUM), Postdoctoral Researcher | Christian Gehrmann, Maximilian Schilcher, Xiangzhou Zhu (TUM), Doctoral Candidates
(Image: Andreas Heddergott)
It is exciting to explore the “wiggling and jiggling of atoms,” as famously coined by Richard Feynman, in the molecules and materials all around us, because virtually every physical and chemical process depends on how atoms are moving about. In energy materials, which are important components in devices for energy conversion or storage such as solar cells or batteries, atomic motions at higher temperatures are important to understand when we want to make these technologies more efficient or less expensive. This is because novel types of energy materials, such as halide perovskites (HaPs), exhibit intriguing atomic motions that deviate from the canonical, wavelike oscillations found in materials such as diamond or silicon. Instead, the atomic behavior is more anharmonic and perturbs the crystal in ways that are not fully understood today. The goal of this Focus Group, which was established in 2019, is to develop and apply theoretical models in order to characterize these motions and their consequences for energy materials on the atomic scale.
Figure 1
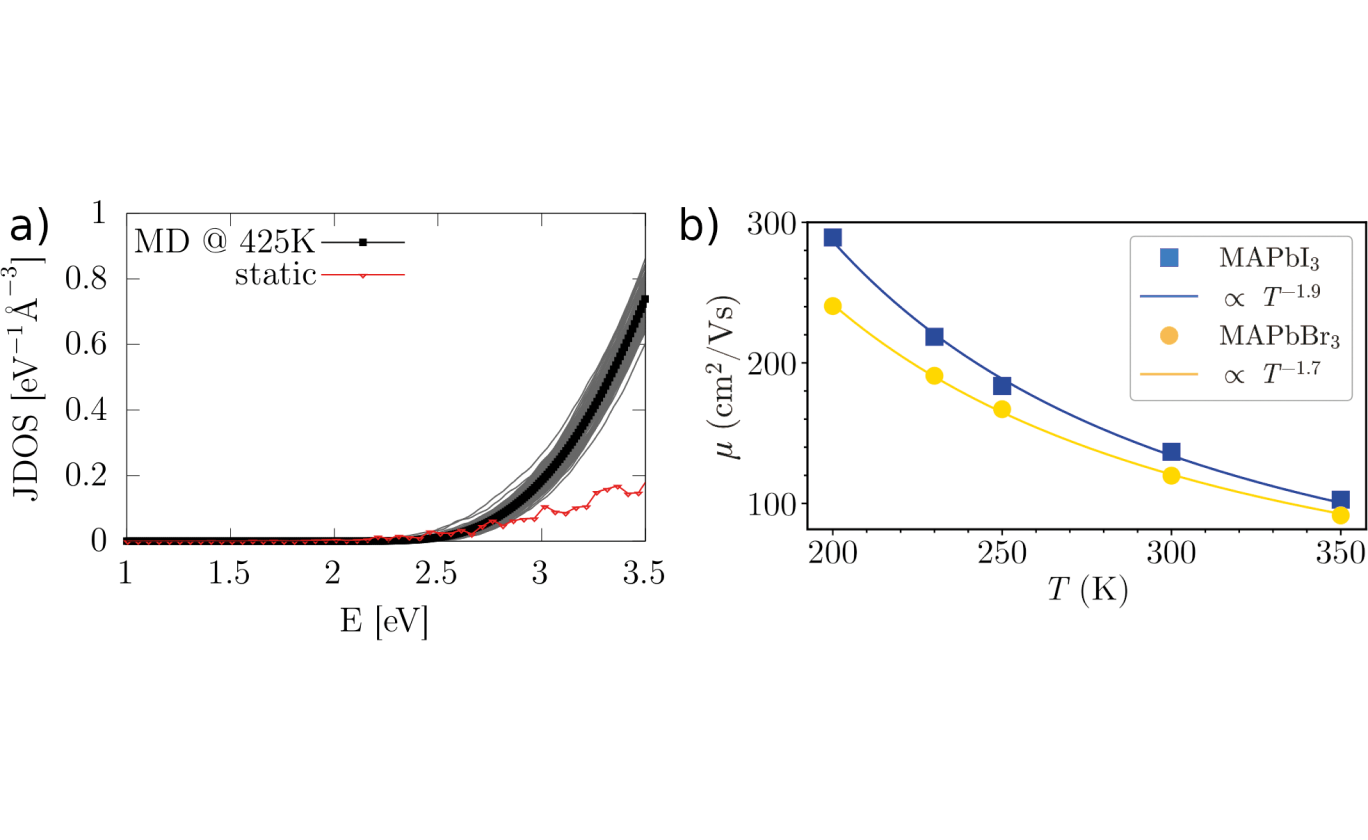
(b) Temperature-dependent charge transport in MAPbI3 and MAPbBr3: carrier mobilities computed in a multi-scale tight-binding model and quantum dynamics. The lines represent fits to the power-law behavior of the temperature-dependent data. (Phys. Rev. Materials 7, L081601 (2023))
Research focus and progress
Since its establishment, the Focus Group has assembled a team of approximately 15 members from nine different countries. Their research work has been published in close to 30 articles. In the following, examples from two research areas, namely on HaPs and solid-state ionic conductors (SSICs), will be summarized to showcase how the atomic wiggling and jiggling impact important properties of energy materials.
HaPs are promising materials for photovoltaics: Solar cells made from them have already surpassed the 25 % power-conversion efficiency threshold, which is the single most important quantity for technological exploitation of photovoltaics. What is more, combination of HaPs with silicon in so-called tandem devices is getting close to reaching 35 % efficiency. In our group, we study the fundamental properties of these materials using quantum-mechanical calculations.
Fig. 1a shows how atomic motions influence key characteristics of HaPs: the joint density of states, which determines how efficiently the material can absorb sunlight, is found to rise significantly more steeply when the atoms move instead of standing still. In Fig. 1b, temperature-dependent carrier mobilities are reported. These are key quantities because they tell how easy it is to transport charge throughout the crystal. Developing a new multi-scale model, we found that anharmonic atomic motions are correlated with carrier transport in HaPs. Switching the composition from MAPbI3 to MAPbBr3 we were able to tune carrier-transport characteristics through these phenomena. This exemplifies how a better understanding of dynamic effects can lead to new ways of controlling important macroscopic properties of materials.
In regard to atomic motions, SSICs share several characteristics with HaPs and other related energy materials. A key difference, however, is that long-range ion migration occurs as the defining feature of SSICs through, e.g., a diffusion mechanism. When SSICs are exploited technologically in new types of batteries, it is crucial to improve the conductivity of mobile ionic species while balancing other material parameters such as the energy density or cost. Our group is interested in revealing how the complex dynamic atomic properties in these materials impact their ionic conductivity.
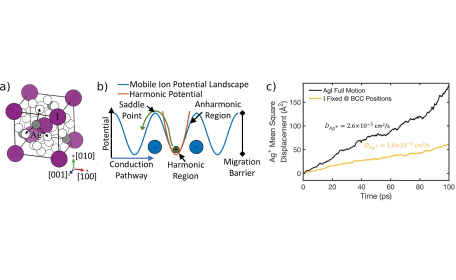
We investigated the perhaps best known SSIC, namely AgI in its superionic α-phase (see Fig. 2a). In this phase, the material shows a high ionic conductivity and the Ag+ ion dynamically hops among various sites. When this occurs, the mobile ionic species ought to enter a potential-energy region of the material that is significantly anharmonic (see Fig. 2b). Together with our experimental partners (group of Dr. Yaffe at the Weizmann Institute of Science, Israel), we showed that the host lattice surrounding the mobile ions plays an important role in the conduction process. Specifically, the anharmonic vibrational coupling among mobile ions and the ones of the host lattice was found to manifest in an interesting duality of crystalline and liquid behavior in AgI. In our molecular dynamics (MD) calculations, we could show that freezing out the atomic motions of the host lattice significantly reduces the ionic conductivity (see Fig. 2c). More recently, we discovered similar types of phenomena in Na-based SSICs, which are among the most promising materials to augment current battery technologies based on Li and other elements. These findings highlight again how interplay of dynamic effects on the atomic scale determine macroscopic functionalities of important types of energy materials with relevance for technological applications.
Figure 2
Outlook
Atomic-scale modeling has reached an exciting level of sophistication and has become an indispensable tool in the discovery and characterization of new materials. We witnessed an enormous acceleration in the field prompted by the advent of artificial intelligence, data science, and machine learning. In our most recent work, we exploited the great opportunities offered by these methods to develop a new theoretical methodology. It allows for atomistic material modeling under more realistic conditions, treating to the full extent the atomic motions as well as their consequences for important properties. We anticipate that further advances in these directions will eventually bring the theoretical and computational research closer to the experimental and technological reality of materials, e.g., in regard to defects and other crystalline irregularities. Bridging computational modeling to the lab and fab will offer new perspectives for revealing fundamental phenomena in materials and exploiting them in technological applications.
Selected publications
-
Righetto, M., Caicedo-Davila, S., Sirtl, M. T., Lim, V. J.-Y., Patel, J. B., Egger, D. A., Bein, T. & Herz, L. M. Alloying Effects on Charge-Carrier Transport in Silver-Bismuth Double Perovskites. J. Phys. Chem. Lett. 14, 10340 (2023).
-
Schilcher, M. J., Abramovitch, D. J., Mayers, M. Z., Tan, L. Z., Reichman, D. R. & Egger, D. A. Correlated Anharmonicity and Dynamic Disorder Control Carrier Transport in Halide Perovskites. Phys. Rev. Mater 7, L081601 (2023).
-
Gehrmann, C., Caicedo-Dávila, S., Zhu, X. & Egger, D. A. Transversal Halide Motion Intensifies Band-To-Band Transitions in Halide Perovskites. Adv. Sci. 9, 2200706 (2022).
-
Schilcher, M. J., Robinson, P. J., Abramovitch, D. J., Tan, L. Z., Rappe, A. M., Reichman, D. R. & Egger, D. A. The Significance of Polarons and Dynamic Disorder in Halide Perovskites. ACS Energy Lett. 6, 2162 (2021).
-
Brenner, T., Gehrmann, C., Korobko, R., Livneh, T., Egger, D. A. & Yaffe, O. Anharmonic host Lattice Dynamics Enable Fast Ion Conduction in Superionic. AgI. Phys. Rev. Mater 4, 115402 (2020).