Exploring epimutational processes across time scales
The field of epigenetics studies heritable changes in genome function that cannot be explained by alterations in the underlying DNA sequence. Our group is interested in understanding how genetic, environmental, and stochastic factors induce epigenetic modifications in plants, how these modifications are propagated over time, and whether they have agricultural and evolutionary consequences.

Focus Group: Population Epigenetics and Epigenomics
Prof. Frank Johannes (TUM), Alumnus Rudolf Mößbauer Tenure Track Assistant Professor | Prof. Robert J. Schmitz (University of Georgia), Alumnus Hans Fischer Fellow | Yadollah Shahryary (TUM), Doctoral Candidate
(Image: Astrid Eckert)
Heritable gains or losses of cytosine methylation can arise stochastically in plant genomes independently of DNA sequence changes. These so-called “spontaneous epimutations” appear to be a by-product of imperfect DNA methylation maintenance. There is continued interest in the plant epigenetics community in trying to understand the broader implications of these stochastic events, as some have been shown to induce heritable gene expression changes, to shape patterns of methylation diversity within and among plant populations and to be responsive to multi-generational environmental stressors. In collaboration with Hans Fischer Fellow Robert J. Schmitz, a central research aim of our Focus Group has been to study the rate and spectrum of spontaneous epimutations, their molecular origins, and their patterns of somatic and transgenerational accumulation. To that end, we have combined mathematical modeling and high-throughput DNA methylation data with various experimental (epi)mutation accumulation systems (Fig. 1).
Figure 1
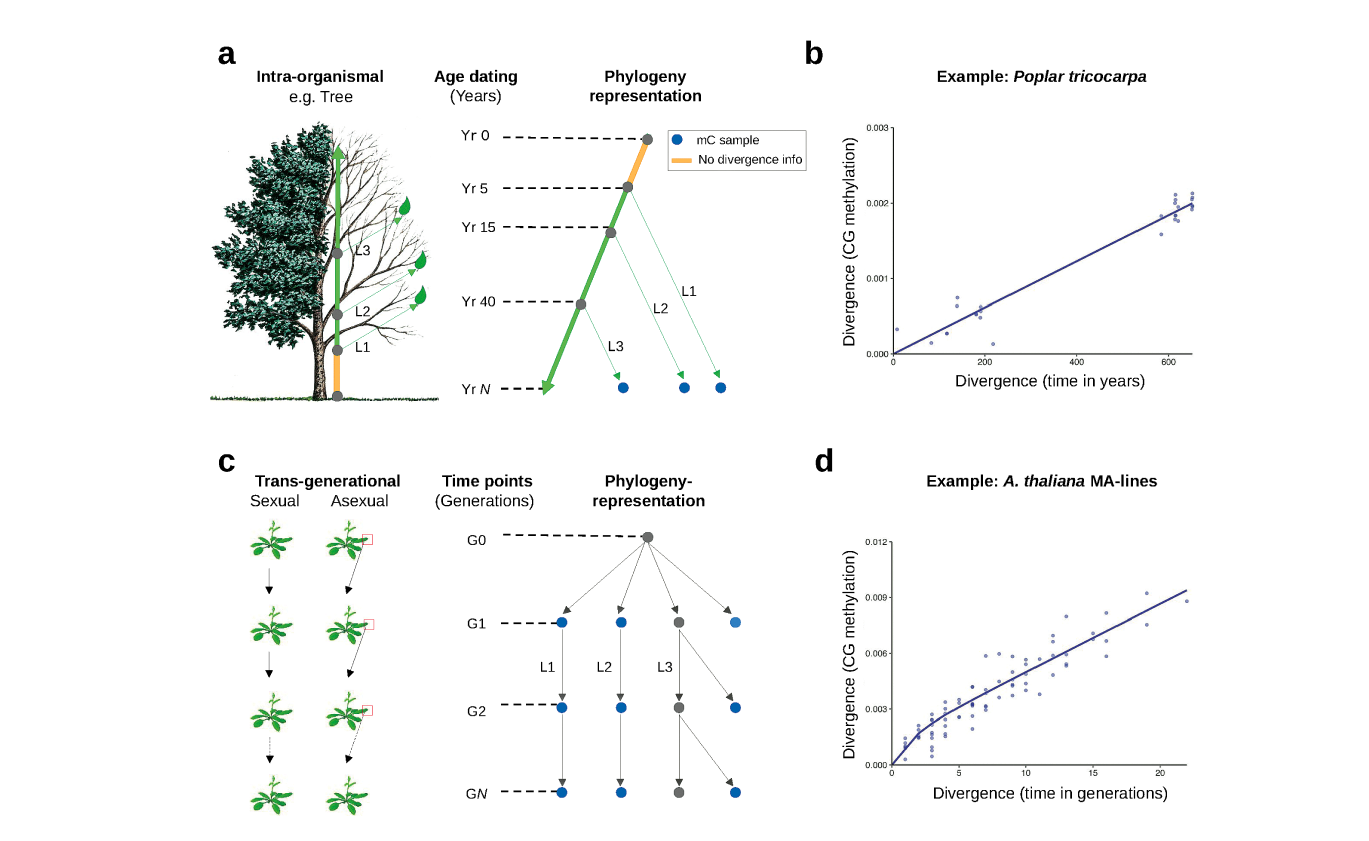
Trends Genet. 37(8), (2021))
Plant genomes harbor epimutation hotspots
Multigenerational studies in plants have shown that the rate of spontaneous epimutations varies substantially across the genome, with some regions harboring localized “epimutation hotspots.” Using the model plant species A. thaliana, we were able to show that epimutation hotspots are indexed by a specific set of epigenomic features that map to a subset of genes. Although these regions comprise only ~12 % of all CGs (cytosine-guanine dinucleotides) in the genome, they account for ~63 % f all epimutation events per unit time. Molecular profiling revealed that these regions contain unique sequence properties, harbor steady-state intermediate methylation levels, and act as putative targets of antagonistic DNA methylation pathways. We further demonstrated that experimentally-induced shifts in steady-state methylation in these hotspot regions are sufficient to significantly alter local epimutation intensities.
Epimutations accumulate during somatic development
Although our work with experimental (epi)mutation accumulation lines (Fig. 1A) has allowed us to gain quantitative insights into the process by which DNA methylation maintenance mistakes arise and are propagated across generations, we actually had no clear understanding of where in the plant life cycle these mistakes originate. One hypothesis was that they occur continuously during somatic development. To test this, we used trees as a model system. Given their exceptional longevity and well-defined modular architectures, trees act as natural (epi)mutation accumulation systems and permit unprecedented insights into the dynamics, mitotic stability, and functional impact of spontaneous epimutations over time scales that have been inaccessible to previous prospective studies. We developed novel analytical methods that treat the tree branching topology as an intra-organism phylogeny and used this approach to show that somatic epimutations accumulate gradually throughout development (Fig. 1B). Moreover, we found that this accumulation was clock-like and that information about somatic epimutations could be used to estimate the chronological age of trees.
Epimutations define a fast-ticking evolutionary clock
Since spontaneous epimutations are also heritable across generations, we sought to test if their clock-like properties could be used, more generally, as a molecular clock to date evolutionary events over longer time scales. In evolutionary biology, DNA-based molecular clocks are classically used for dating the divergence between lineages over macroevolutionary time scales (~105 to 108 years). However, these classical DNA-based clocks tick too slowly to inform us about the recent past. We were able to demonstrate that stochastic DNA methylation changes at a subset of cytosines in plant genomes define an epimutation-based evolutionary clock whose tick-rate is orders of magnitude faster than DNA-based clocks. This new clock enabled us to perform phylogenetic explorations on a scale of years to centuries. We showed experimentally that the use of epimutation clocks recapitulate known topologies and branching times of intraspecies phylogenetic trees in the self-fertilizing plant A. thaliana and the clonal seagrass Z. marina, which represent two major modes of plant reproduction. This discovery will open new possibilities for high-resolution temporal studies of plant biodiversity.
Future research
Our group continues to dissect the molecular and phenotypic properties of spontaneous epimutations in plants. We have begun to employ single-cell sequencing technologies to be able to detect the origin of somatic epimutations within a small population of stem cells from in which they most likely arise. Using cell lineage-based mathematical models, we further seek to understand how somatic epimutations originating in this stem cell population are propagated in the plant morphological architecture throughout development, as well as how they are eventuallly transferred to the cell lineages that form the next generation. In parallel to these efforts, we aim to calibrate epimutation-based evolutionary clocks in various plant species to help resolve evolutionary questions about time scales that are difficult to resolve with DNA-based mutation data.
Selected publications
-
Yao N. et al. An evolutionary epigenetic clock in plants. Science 381, 1440-1445 (2023).
-
Hazarika, R.R. et al. Molecular properties of epimutation hotspots. Nature Plants 8, 146–156 (2022).
-
Zhang Y. et al. Heterochromatin is a quantitative trait associated with spontaneous epiallele formation. Nature Communications 12, 6958 (2021).
-
Shahryary D.Y. et al. AlphaBeta: Computational inference of epimutation rates and spectra from high-throughput DNA methylation data in plants. Genome Biology 21, 260 (2020).
-
Lauss K. et al. Parental DNA methylation states are associated with heterosis in Arabidopsis epigenetic hybrids. Plant Physiology 176, 1627-1645 (2018).