From understanding drug action to treating cancer patients
Most cancer drugs modulate the activity of malfunctioning proteins. However, it is often not clear how these drugs work and the molecular heterogeneity of tumors often render drugs ineffective. The goal of this Focus Group is to provide better treatment options to patients based on a detailed understanding of their tumor biology and exploiting the capabilities of existing targeted cancer drugs.

Focus Group Understanding How Cancer Drugs Work
Prof. Bernhard Küster (TUM), Alumnus Carl von Linde Senior Fellow
(Image: Andreas Heddergott)
Proteins execute and control practically every aspect of life. The human genome encodes for about 20,000 proteins, which, among many other functions, act as catalysts for anabolic or catabolic biochemical reactions inside cells, help to transmit signals via the network of neurons or control cell growth and division. Deregulated proteins can cause severe diseases including heart or liver disorders, neurodegenerative diseases or cancer to name a few. Therefore, most therapeutic drugs act on proteins with the goal to tone down or increase their activity in order to restore normal cellular function.
Despite a very long history of successful drug development, surprisingly little is known about how drugs impact the entirety of the human proteome or vice versa. Drugs sometimes do not work in humans even though the scientific rationale and pre-clinical data is strong. Often, drugs, have more than one effect, a phenomenon called polypharmacology, which may or may not be desirable. In part, this is due to an astounding molecular diversity of proteins in different cell types or organs and there is also strong heterogeneity in composition and dynamics of the proteome between (cancer) patients.
Understanding the complex interplay between a therapeutic compound and the cellular proteome and how this information may be used to make better choices for the treatment of cancer patients is the goal of this Focus Group. Our unique approach to this challenge builds on one of the world’s most powerful proteomic technology platforms established by an interdisciplinary team of molecular oncologists, biochemists and bioinformaticians. A few highlights from published and ongoing work are provided in the following:
1. From our analysis of the human and mouse proteomes (Figure 1) [1], as well as hundreds of cancer patient proteomes, it has become very clear, that while the majority of all proteins is expressed in most organs or cancers, their quantities in these organs can be vastly different. This is even more pronounced for the many chemical modifications with which proteins can be decorated and which strongly regulate their function. It should, therefore, no longer be surprising that drugs will encounter very different molecular environments when entering a particular cell.
Figure 1

2. We and others have screened hundreds of cancer cell lines against hundreds of cancer drugs to measure the ability of drugs to kill these cells [2]. Reflecting the aforementioned heterogeneity of cancer proteomes, most cancer drugs do not work in most cancer cells. Still, using advanced bioinformatic analysis, we were able to show that the expression of certain proteins, or the levels of phosphorylation modification on certain proteins, are predicative of response or resistance to certain cancer drugs. We exemplified the utility of such biomarkers of drug response by i) improving the stratification of breast cancer patients for anti-hormonal therapy and ii) suggesting higher doses of Cytarabine for the treatment of acute myeloid leukemia patients with high expression levels of the protein Adenylate Kinase 1.
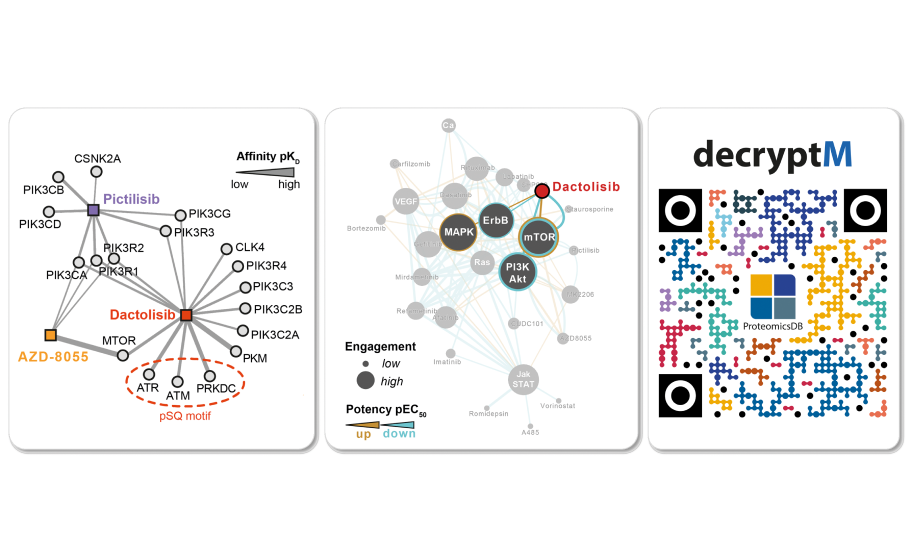
3. One key element in understanding how drugs work is to identify the target proteins they engage and we have investigated hundreds of cancer drugs (mostly kinase inhibitors (KI) and histone deacetylation inhibitors (HDACi) for their ability to engage cellular proteins and pathways (Figure 2) [3]. This work not only provided systematic information on drug:target interactions, it also uncovered several unexpected targets that could only be discovered in a proteome-wide approach. The biological function of some of these so-called “off-targets” are not yet clear but for others, these additional target proteins explained toxic side effects of the drug, or suggest additional uses of the same drug for a new target and in new indications (a concept known as drug re-purposing). The same line of work also identified the targets of the drug lipoic acid which has been used in humans for decades in the absence of knowing how it exerts its therapeutic effects.
4. In ongoing work, we have extended our efforts to the direct measurement of the phospho-proteome of cancer cells in response to cancer drugs. The scale of these experiments is unprecedented and we have already accumulated millions of dose-response measurements with the aim to understand the details of the cellular mechanism of action (MoA) of these drugs. Important learnings have emerged from this work already. For instance, we could show that the classic breast cancer drug Herceptin is not a signaling drug. We could also demonstrate that the therapeutic antibody Rituximab kills cancerous B-cells by overactivating a particular intracellular signaling pathway, thus clarifying the MoA of a 25 year old and successful drug.
Figure 2
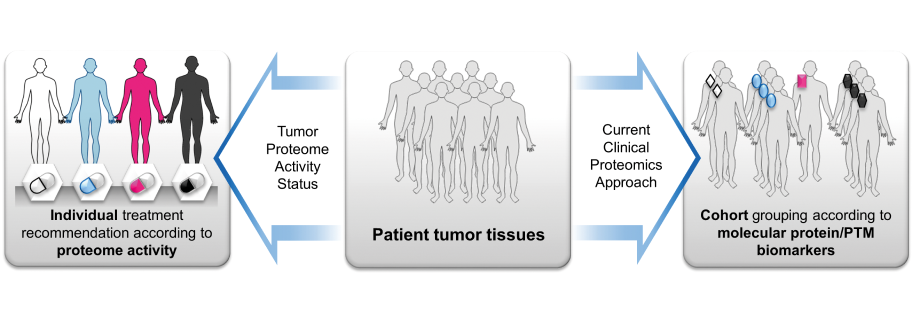
All the above research areas serve a common long-term goal: to provide better treatment options to cancer patients based on a detailed understanding of their individual tumor biologies and the molecular capabilities of molecularly targeted cancer drugs (Figure 3). We are in the process of pulling together the above lines of work as part of an ERC Advanced Grant as well as our participation in the German Consortium for Cancer Research (DKTK). We have already measured the phosphoproteomes of hundreds of cancer patients and these are analyzed and thousands more are to come. This will place the molecular profile of every patient into context of tumor heterogeneity and identify features that are characteristic for this particular patient. We then pair this information with data for the cancer drugs regarding which of an aberrantly regulated protein in a patient, matches the modulation profile of a drug. We have already begun to use this concept for treatment recommendations in molecular tumor boards and will incorporate it in retrospective and prospective clinical trials in order to learn, if the proteomic profiles and recommendations derived provide any benefit to the lives of patients.
In close collaboration with Prof. Dr. Mathias Wilhelm, Dr. Maria Reinecke, Dr. Matthew The, Dr. Stephanie Wilhelm, Dr. Patrick Allihn, Dr. Chen Meng, Dr. Chien-Yun Lee, Dr. Guillaume Medard, Dr. Jana Zecha, Dr. Patroklos Samaras, Dr. Svenja Wiechmann, Dr. Martin Frejno, Florian Bayer, Julian Müller, Nicola Berner, Nicole Kabella, Severin Lechner, Stefanie Höfer, Stephan Eckert, Yun-Chien Chang (TUM).
Figure 3
[1]
Wilhelm, M. et al. Mass spectrometry based draft of the human proteome. Nature 509, 582–587 (2014); Giansanti, P. et al. (2022).
[2]
Frejno, M. et al. (2020).
[3]
Klaeger, S. et al. The target landscape of clinical kinase drugs. Science 358, pii: eaan4368 (2017); Lechner, S. et al. (2022).
Selected publications
-
Lechner, S. et al. Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common off-target. Nat Chem Biol 18, 812–820 (2022), doi.org/10.1038/s41589-022-01015-5.
-
Giansanti, P. et al. Mass spectrometry-based draft of the mouse proteome. Nat Methods 19, 803–811 (2022), doi.org/10.1038/s41592-022-01526-y.
-
Frejno, M. et al. Proteome activity landscapes of tumor cell lines determine drug responses. Nat Commun 11, 3639 (2020), doi.org/10.1038/s41467-020-17336-9.
-
Jarzab, J. et al. Meltome Atlas – thermal proteome stability across the tree of life. Nat Methods 17, 495–503 (2020), doi.org/10.1038/s41592-020-0801-4.
-
Reinecke, M. et al. Chemoproteomic selectivity profiling of PIKK and PI3K kinase inhibitors. ACS Chemical Biology 14, 655–664 (2019), doi.org/10.1021/acschembio.8b01020.