Investigating spontaneous epigenetic variation
Genetic variation is a major driver of trait variation. Missing from previous efforts is an understanding of how epigenetic variation contributes to this process. We focus on innovating methods, studying experimental and natural plant populations, and developing mathematical models to better understand the role of epigenetic variation in trait variation.

Focus Group Population Epigenetics and Epigenomics
Prof. Robert Schmitz (University of Georgia), Alumnus Hans Fischer Fellow | Yadollah Shahryary (TUM), Doctoral Candidate | Host: Prof. Frank Johannes (TUM)
We study how chemical modification to the DNA sequence (DNA methylation) varies naturally in plants. It is well known that some of these changes lead to turning off the gene, whereas others currently have no known function. Much of the effort in this area is focused on how development or the environment affects these chemical modifications. Our group is unique in that we specifically focus on spontaneous changes to DNA methylation that accumulate naturally. Our work has demonstrated that spontaneous changes to DNA methylation accumulate over cell division and generational time, yet the origins of these changes remain unknown. Additionally, we study if there are any functional consequences associated with these spontaneous epigenetic changes. Finally, our discoveries that spontaneous epigenetic variation accumulates with time and at a rate much higher than the spontaneous DNA mutation rate provides a unique opportunity use spontaneous epigenetic variation as a molecular clock.
To better understand the role of spontaneous epigenetic variation, we use both natural and experimental plant populations ranging from plants that have short annual life styles to those that are long-lived perennials. The natural life history of each plant species provides an opportunity to evaluate how it influences the rate and spectrum of spontaneous epigenetic variation. Our studies can be classified into two distinct classes: 1) studies of natural epigenetic variation and 2) studies of experimentally induced epigenetic variation.
- Natural epigenetic variation: We initiated a survey of natural spontaneous changes in DNA methylation in a collection of more than 750 wild isolates of Arabidopsis thaliana. Each of these isolates represents a different individual of a genetically distinct population. We characterized the DNA methylation status of genes into three categories: unmethylated, gene body DNA methylated, and genes that had transposon / repeat-like methylation patterns. We observed that the vast majority of genes were consistently in an unmethylated state, regardless of the geographical origin. A rare fraction of genes was present in transposon / repeat-like methylation state. Approximately 15 % of genes were classified as gene body DNA methylated, and this class exhibited the greatest amount of variation in genes between different wild isolates. We explored the basis for this variation and observed a unique pattern that suggests that the variation observed is associated with DNA methylation pathways that were primarily thought to function only in transposon / repeat regions of plant genomes. This research has opened a new path of research to better understand how these distinct regions of the genome interact to lead to spontaneous epigenetic variation.
- Experimentally induced variation: The major breakthrough that we made prior to beginning this Fellowship was that spontaneous epigenetic variation accumulates over time. This conclusion was based on the use of A. thaliana mutation accumulation lines where experimental populations of single seed descent lineages were created from a single founder individual. This experimental setup allowed us to control the known relationship and time between parent and progeny individuals.
Figure 1
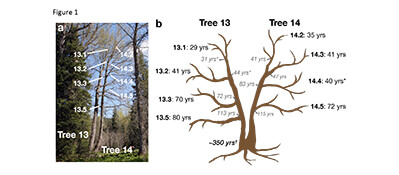
(a) Leaf samples were collected from the labeled terminal branches.
(b) Age was estimated for both the end of the branch (black font) and where it meets the main stem (gray italics). Ages with * indicate age was estimated using diameter; all other estimates were from core samples.
One major hypothesis is that that spontaneous epigenetic variation forms as a by-product of reproduction when DNA is inherited from one generation to the next. To test this hypothesis, we isolated DNA from eight different branches of a long-lived perennial poplar tree that is approximately 350 years old. This experimental setup allows us to explore if spontaneous epigenetic variation accumulates with time independent of sexual reproduction. We were able to trace back the accumulation of changes to DNA methylation to a common ancestral cell and determine the extent of spontaneous DNA methylation variation. An important discovery was that these variations in DNA methylation exist even though none of the samples were collected from across generations. These results show that spontaneous changes to DNA methylation, which sometimes are linked to gene expression and trait variation, accumulate as a result of cells replicating their DNA within generations and not between generations.
In parallel, we have designed additional mutation accumulation populations whereby we have used founder plants that are mutant in key enzymes we hypothesize influence the rate and spectrum of spontaneous epigenetic variation. At the same time, have leveraged our expertise in plant epigenomics to better understand features that might underlie why certain regions of plant genomes are more susceptible than others to spontaneous epigenetic variation. Combining these two approaches has led to the discovery that a minor portion of plant genomes contributes a major source of epigenetic variation, and this allows us to measure divergence time between individuals where not enough DNA mutations have done so to make accurate predictions of relatedness.
A number of major discoveries have resulted from this unique collaboration.
- The epimutation rate is a product of cell division time and is independent of major epigenetic reinforcement strategies that plants use during sexual reproduction.
- Epimutation rates are stable and predictable enough that they can be used to estimate age of individuals, providing a new molecular clock. This is especially powerful for use in situations in which there has not been enough time for DNA mutations to accumulate to a point where they are useful in determining relatedness and divergence time between individuals.
- We have discovered that DNA methylation pathways important for silencing transposons and repeats are important for creating the spontaneous epigenetic variation observed in genes.
- We have determined the molecular epigenomic properties of regions of the genome that are most susceptible to accumulation of spontaneous epimutations.
These results have led to a major breakthrough: We are confident that epimutation rates can be used to age-date individuals in plant populations in cases where there is not enough DNA sequence variation to do so. This has major implications for studying speciation and invasiveness, for mapping of relationships between clonally propagated plants, and for age-dating plants.
Our future research will explore how the epimutation varies between species and determine how applicable these epimutation models can be when used to evaluate divergence time between individuals. In parallel, we will continue our studies aimed at understanding how epimutations originated. We firmly believe understanding this process will provide clues as to why their functions exist in plants, which is still a mystery.
Selected publications
- Wendte, J. M., Zhang, Y., Ji, L., Shi, X., Hazarika, R. R., Shahryary, Y., Johannes, F. & Schmitz, R. J. Epimutations are associated with CHROMOMETHYLASE 3-induced de novo DNA methylation. eLife 8:e47891 (2019), https://doi.org/10.7554/eLife.47891.
- Hofmeister, B. T., Denkena, J., Colom.-Tatch., M., Shahryary, Y., Hazarika, R., Grimwood, J., Mamidi, S., Jenkins, J., Paul P. Grabowski, P. P., Avinash Sreedasyam, A., Shengqiang Shu, S., Barry, K., Lail, K., Adam, C., Lipzen, A., Sorek, R., Kudrna, D., Talag, J., Wing, R., Hall, D. W., Jacobsen, D., Tuskan, G. A., Schmutz, J., Johannes, J. & Schmitz, R. J. A genome assembly and the somatic genetic and epigenetic mutation rate in a wild long-lived perennial Populus trichocarpa. Genome Biol. 21, Art. no. 259 (2020), https://doi.org/10.1186/s13059-020-02162-5.
- Shahryary, Y., Symeonidi, A., Hazarika, R. R., Denkena, J., Mubeen, T., Hofmeister, B., van Gurp, T., Colom.-Tatch., M., Verhoeven, K. J. F., Tuskan, G., Schmitz, R. J. & Johannes, F. AlphaBeta: Computational inference of epimutation rates and spectra from high-throughput DNA methylation data in plants. Genome Biol. 21, Art. no. 260 (2020), https://doi.org/10.1186/s13059-020-02161-6.
- Zhang, Y., Jang, H., Xiao, R., Kakoulidou, I., Piecyk, R. S., Johannes, F. & Schmitz, R. J. Heterochromatin is a quantitative trait associated with spontaneous epiallele formation. Nat. Commun. 12, Art. no. 6958 (2021), https://doi.org/10.1038/s41467-021-27320-6.
- Hazarika, R. R., Serra, M., Zhang, Z., Zhang, Y., Schmitz, R. J. & Johannes, F. Molecular properties of epimutation hotspots. Nat. Plants 8, 146–156 (2022), https://doi.org/10.1038/s41477-021-01086-7.
Full list of publications